



Rechercher la grâce : poursuivre les demandes d'indemnisation liées au traitement à la lumière des divulgations relatives aux essais cliniques

Cet article a été initialement publié dans le lettre d'information AIPLA Chemical Practice Chronicles Fall 2025 et est republié ici avec autorisation.
I. Contexte
Les brevets de méthode de traitement basés sur les protocoles d'essais cliniques de phase II et III sont systématiquement recherchés afin de prolonger l'exclusivité du brevet et de constituer stratégiquement un portefeuille de brevets pour un actif pharmaceutique. Les revendications de ces brevets de méthode de traitement « de dernière génération » décrivent les caractéristiques saillantes du protocole d'essai clinique de phase II ou III, notamment les populations de patients, les doses, les schémas posologiques et les mesures des résultats d'efficacité. Cela se justifie, car les caractéristiques saillantes du protocole d'étude figurent souvent sur l'étiquette du médicament, parfois dans le cadre de mesures actives explicites.
Pour une nouvelle entité chimique (NCE), un brevet de méthode de traitement de dernière génération offre une durée de validité supplémentaire, parfois supérieure de plusieurs années à celle des brevets de première génération, c'est-à-dire les brevets fondamentaux garantissant l'exclusivité de la composition de la matière et/ou de la méthode de traitement au sens large.
Pour les médicaments réutilisés ou les nouveaux protocoles de dosage, les brevets sur les méthodes de traitement basés sur les protocoles d'essais cliniques de phase II ou III peuvent fournir la seule source significative d'exclusivité, par exemple si le composé est connu et que le brevet sur la composition de la matière a expiré ou est sur le point d'expirer.
La sagesse conventionnelle dicte de déposer une demande de brevet sur la base d'un protocole d'essai clinique de phase II ou III avant la divulgation publique de l'étude afin d'éviter la création d'un état de la technique susceptible d'empêcher la brevetabilité des revendications relatives au procédé de traitement, en particulier en dehors des États-Unis.[1] Compte tenu de l'importance stratégique des brevets de méthode de traitement de dernière génération, il convient de bien comprendre le calendrier des divulgations publiques relatives à l'essai clinique et de planifier le dépôt des demandes de brevet en conséquence.
Une telle divulgation publique liée à un essai clinique consiste à publier le protocole d'essai clinique de l'innovateur sur le site ClinicalTrials.gov (« CTG ») sous la forme d'un « dossier d'étude ».[2] La publication du dossier d'étude s'inscrit dans le cadre des efforts bien intentionnés des États-Unis pour mener des essais cliniques sur l'homme dans la transparence, entre autresrenforcer la confiance du public dans la recherche clinique et aider les patients à trouver les essais auxquels ils pourraient être éligibles. Les innovateurs doivent soumettre les protocoles d'essais cliniques à la FDA au plus tard 21 jours après l'inscription du premier patient à l'essai[3], et le dossier de l'étude sera publié au plus tard 30 jours après la soumission[4][5. Compte tenu de ces délais, il n'est généralement pas possible de prédire le jour exact où un dossier d'étude sera rendu public sur le CTG. Un tel délai « non enregistrable », contrairement à la date fixe de publication d'un article dans une revue ou d'un symposium public, a conduit, sans surprise, à une situation où un dossier d'étude est publié sur le CTG avant le dépôt d'une demande de brevet pour la méthode de traitement correspondante.
II. Recours à l'exception prévue au § 102(b)(1)(A)
Les essais cliniques eux-mêmes ne sont pas considérés comme une utilisation publique antérieure au sens de l'article 35 U.S.C. § 102(a)(1).[6] Les dossiers d'études CTG et autres divulgations liées aux essais cliniques sont toutefois considérés comme des publications imprimées et/ou « autrement accessibles au public » en vertu des §§ 102(a)(1) s'ils ont été réalisés avant la date effective de dépôt de la demande de brevet. Ces divulgations liées aux essais cliniques peuvent donc être utilisées pour rejeter les revendications relatives à la méthode de traitement d'une demande de brevet déposée ultérieurement, car elles sont anticipées au sens de l'article 35 U.S.C. § 102(a)(1) et/ou évidentes au sens de l'article 35 U.S.C. § 103.
Dans le cas où un dossier d'étude CTG ou une divulgation connexe serait rendu public avant le dépôt d'une demande de brevet américain correspondant pour une méthode de traitement, l'exception relative au délai de grâce prévue à l'article 102(b)(1)(A) pourrait être invoquée pour invalider la divulgation en vertu de l'article 102(a)(1) – à condition que la divulgation ait été faite un an ou moins avant la date effective de dépôt de la demande. Sur le plan procédural, si un examinateur rejette une demande en vertu des articles 35 U.S.C. §§ 102(a)(1) et/ou 103 sur la base d'un dossier d'étude CTG ou d'une divulgation connexe, le demandeur peut invoquer le délai de grâce en soumettant une déclaration en vertu de l'article 37 CFR § 1.130 (« règle 130 ») établissant que la divulgation a été faite par l'inventeur ou les co-inventeurs et demandant le retrait du dossier d'étude CTG ou de la divulgation connexe en tant qu'état de la technique.
Bien que la soumission de la déclaration prévue à la règle 130 semble simple, un exemple récent du PTAB, Murray & Poole Enterprises Ltd. c. Institut de Cardiologie de Montréal[7] illustre bien les pièges potentiels. L'Institut de Cardiologie de Montréal (ICM) a tenté de supprimer « Bouabdallaoui » en tant qu'état de la technique en invoquant l'exception du délai de grâce prévue à l'article 102(b)(1)(A). Bouabdallaoui a été publié pendant le délai de grâce d'un an et a développé les résultats de l'étude CTG « COLCOT ». Bouabdallaoui mentionnait sept auteurs, dont le deuxième, le Dr Tardif, était le seul inventeur du brevet de l'ICM. La commission a précisé que l'invocation du délai de grâce dépendait de la question de savoir si la déclaration du Dr Tardif fournissait suffisamment d'informations pour conclure que Bouabdallaoui n'était pas « par un autre ».[8] La déclaration du Dr Tardif expliquait la relation de travail avec un coauteur (Bouabdallaoui), comme entre autresreconnaissance de l'aide apportée par Bouabdallaoui dans la conduite de l'essai clinique et pour lui permettre d'être le premier auteur de la publication. La déclaration du Dr Tardif décrivait également la portée de l'essai clinique multicentrique COLCOT et le statut de l'ICM en tant que promoteur, étayé par des accords entre certains des centres et le promoteur. Le comité a souligné que seule une partie des accords entre les centres et le promoteur avait été fournie et que, parmi les accords fournis, aucun ne mentionnait le Dr Tardif comme chercheur principal.[9] En fin de compte, dans sa décision d'ouverture de la procédure, la commission a estimé que la déclaration du Dr Tardif était insuffisante pour disqualifier Bouabdallaoui en tant qu'état de la technique.[10]
Comme Murray , toutes les différences entre les auteurs de la divulgation liée à l'essai clinique et les inventeurs figurant dans la demande de brevet doivent être expliquées de manière exhaustive. Les déclarations de tout auteur superflu, c'est-à-dire non inventeur, renonçant à sa contribution à l'objet invoqué pour rejeter l'état de la technique peuvent aider à établir que la divulgation liée à l'essai clinique n'est pas « par un autre ». Contrairement aux publications classiques, les dossiers d'étude du CTG ne mentionnent que le promoteur d'un essai clinique, et non les personnes spécifiques responsables de la conception de l'étude. Néanmoins, le ou les inventeurs peuvent attester dans une déclaration que le dossier d'étude du CTG est leur propre travail. Des pièces justificatives établissant clairement le lien entre le ou les inventeurs et le dossier d'étude du CTG, par exemple en désignant le ou les inventeurs comme chercheurs principaux, peuvent également être fournies.
III. Dispositions internationales relatives au délai de grâce et stratégies de dépôt
Outre les États-Unis, plusieurs juridictions étrangères accordent également un délai de grâce. Sans être exhaustif, le tableau 1 résume les délais de grâce accordés dans les juridictions étrangères les plus courantes, les délais de dépôt des demandes et la nécessité ou non de prendre des mesures proactives pour bénéficier du délai de grâce. Les praticiens doivent travailler en étroite collaboration avec des avocats locaux afin de comprendre les subtilités des exigences nationales et les délais à respecter pour bénéficier correctement du délai de grâce accordé par chaque pays.
Tableau 1
Pays | Délai de grâce disponible | Délai pour déposer une demande (à compter de la divulgation) | Type de première demande à déposer | La première demande peut-elle servir de documentprioritaire…† | Notes |
|---|---|---|---|---|---|
UA | Oui | 12 mois | Demande standard AU ou PCT | Oui (pour le PCT) | Aucune mesure proactive n'est requise. |
BR | Oui | 12 mois | Provisoire américain, BR ou PCT | Oui | Aucune mesure proactive n'est requise. |
CA | Oui | 12 mois | CA ou PCT | Non | Aucune mesure proactive n'est requise. |
CN | Non | — | — |
| La divulgation CTG ne donne pas droit à la période de grâce. |
EP | Non | — | — |
| La divulgation CTG ne donne pas droit à la période de grâce. |
JP | Oui | 12 mois | JP ou PCT | Non | Dans les 30 jours suivant le dépôt de la demande nationale JP ou l'entrée dans la phase nationale JP, un certificat décrivant plusieurs caractéristiques de la divulgation publique doit être déposé. |
KR | Oui | 12 mois | KR ou PCT | Oui, mais uniquement si le dépôt prioritaire est (a) une demande PCT désignant uniquement la Corée ou (b) une demande directe en Corée. | Des documents probants doivent être présentés pour prouver la divulgation par le demandeur/inventeur et indiquer (i) la date et le type de divulgation, (ii) la partie divulgatrice et (iii) le contenu de la divulgation. |
MX | Oui | 12 mois | Provisoire américain, MX ou PCT | Oui | La date de la divulgation publique doit être indiquée sur un formulaire lors du dépôt de la demande nationale MX ou de l'entrée en phase nationale MX du PCT. |
PH | Oui | 12 mois | Provisoire américain, PH ou PCT | Oui | Aucune mesure proactive n'est requise. |
SG | Oui | 12 mois | PCT | Non | Aucune mesure proactive n'est requise. |
TW | Oui | 12 mois | TW | Non | Aucune mesure proactive n'est requise. |
États-Unis | Oui | 12 mois | Provisoire américain ou PCT | Oui | Aucune mesure proactive n'est requise. |
† pour une demande PCT ou autre déposée après le délai de grâce qui bénéficie du délai de grâce ?
À l'instar des États-Unis, l'Australie, le Brésil, le Canada, le Japon, le Mexique, les Philippines, la Corée du Sud, Singapour et Taïwan accordent tous des délais de grâce pour les dossiers d'études CTG et les divulgations connexes dans les 12 mois suivant la date de dépôt de la première demande de brevet. Ils diffèrent toutefois quant à la question de savoir si la première demande de brevet peut servir de document de priorité pour une demande déposée ultérieurement, par exemple une demande PCT, lorsque la demande nationale de la demande PCT sera également bénéficier du délai de grâce. Certaines juridictions, par exemple la Chine et l'Europe, n'accordent en fait aucun délai de grâce pour les dossiers d'études sur les CTG ou les divulgations connexes.
Compte tenu de ces distinctions, on peut envisager des stratégies de dépôt complexes en fonction des priorités internationales en matière de dépôt.
Le scénario 1 est une stratégie de dépôt « classique » dans laquelle une demande provisoire américaine est la première demande déposée dans la famille. Cette stratégie de dépôt peut être suivie pour obtenir une couverture par un brevet dans les pays où une demande provisoire américaine déposée dans les 12 mois suivant l'enregistrement de l'étude CTG ou la divulgation connexe peut servir de document de priorité pour une demande PCT déposée 12 mois après la demande provisoire américaine, et les demandes en phase nationale de la demande PCT peuvent bénéficier du délai de grâce basé sur la date de dépôt de la demande provisoire américaine. Les pays représentatifs dans lesquels cette stratégie de dépôt peut être utilisée comprennent les États-Unis, le Brésil, le Mexique et les Philippines.
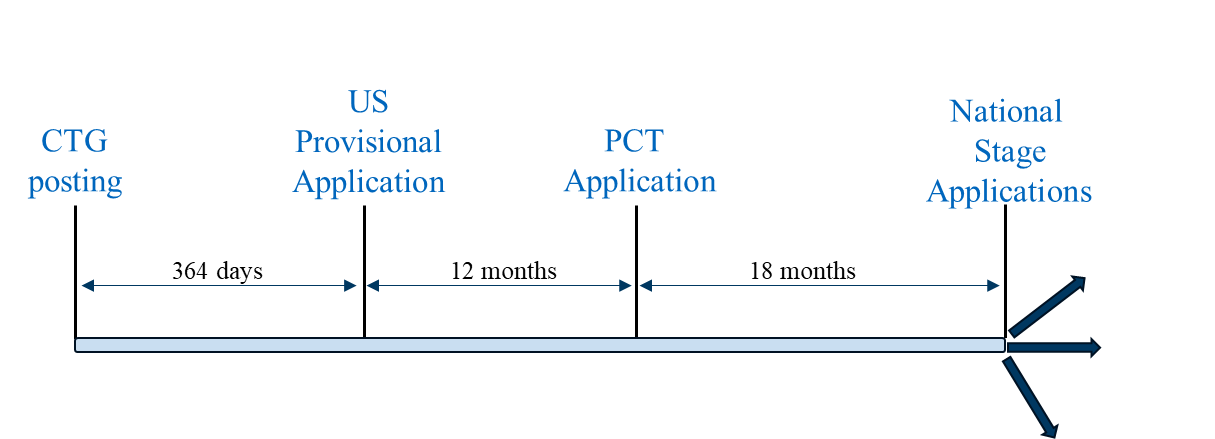
Scénario de classement 1
Le scénario 1 peut également être suivi dans les pays qui ne disposent pas réellement ou effectivement de délais de grâce (par exemple, la Chine et l'Europe). Pour surmonter l'obstacle de la nouveauté absolue en Chine, les aspects éventuels des méthodes commerciales prévues qui ne sont pas divulgués dans la divulgation relative à l'essai clinique peuvent être inclus dans la demande provisoire américaine et/ou la demande PCT. En Europe, les dossiers d'études du CTG ou les annonces concernant les essais en cours ne sont pas considérés comme détruisant la nouveauté, bien qu'ils puissent être utilisés dans une analyse de l'activité inventive.[11] Pour obtenir des conseils sur les implications des essais cliniques en tant qu'état de la technique en Europe, nous renvoyons le lecteur à l'article informatif du Comité des pratiques chimiques de l'AIPLA du printemps 2024 sur ce sujet précis.[12]
Le scénario 2 peut être suivi dans les pays où une demande nationale PCT ou non PCT doit être déposée dans un délai d'un an à compter de la divulgation publique pour que la demande nationale puisse bénéficier d'un délai de grâce. Les pays représentatifs dans lesquels cette stratégie peut être utilisée sont notamment l'Australie, le Canada, le Japon, la Corée, Singapour et Taïwan.
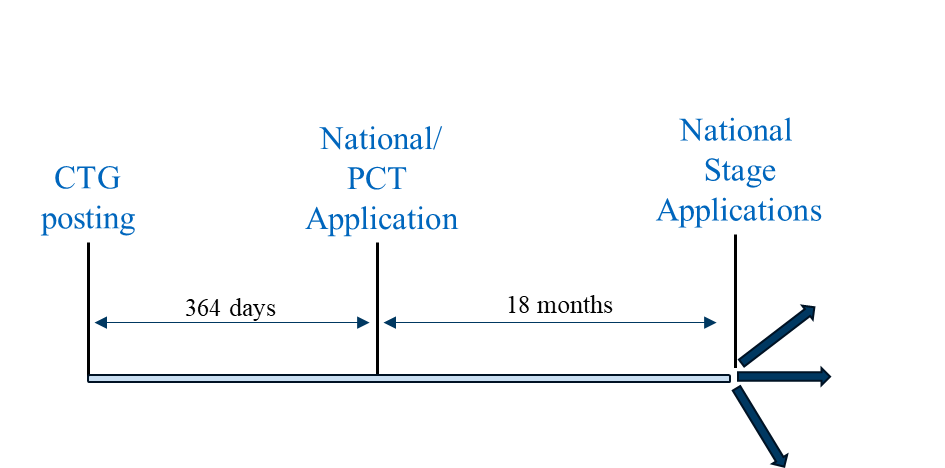
Scénario de classement 2
Le scénario 1 offre deux avantages potentiels par rapport (a) à une stratégie de dépôt plus conservatrice, dans laquelle la demande provisoire américaine est déposée avant la publication du dossier d'étude CTG, et (b) au scénario 2 : (1) un délai maximal pour obtenir les résultats des essais cliniques qui, s'ils sont disponibles, peuvent être inclus dans la demande PCT, et (2) la possibilité d'obtenir une durée maximale pour le brevet.
Le scénario 2 n'offre pas l'avantage d'une durée prolongée entre la publication du rapport d'étude CTG et le dépôt d'une demande nationale ou PCT. Ainsi, les résultats de l'essai clinique sont généralement moins susceptibles d'être disponibles au moment du dépôt d'une demande PCT ou d'une demande nationale dans un pays non PCT. Cependant, bon nombre des pays susmentionnés autorisent la présentation de données postérieures au dépôt pendant la procédure, ce qui permet de présenter les résultats des essais cliniques.[13]
Le scénario 3 combine les scénarios 1 et 2 en une stratégie de dépôt unifiée. Tout d'abord, une demande provisoire aux États-Unis (conformément au scénario 1), une première demande PCT (PCT1) (conformément au scénario 2) et une ou plusieurs demandes nationales non PCT (conformément au scénario 2) sont déposées le même jour et dans un délai d'un an à compter de la publication du dossier d'étude du CTG. Avant de déposer toute demande en dehors des États-Unis, il convient d'envisager l'obtention d'une licence de dépôt à l'étranger et, si nécessaire, de l'obtenir. La demande PCT1 peut être nationalisée dans les juridictions où les exigences en matière de délai de grâce sont plus strictes, comme indiqué ci-dessus pour le scénario 2.
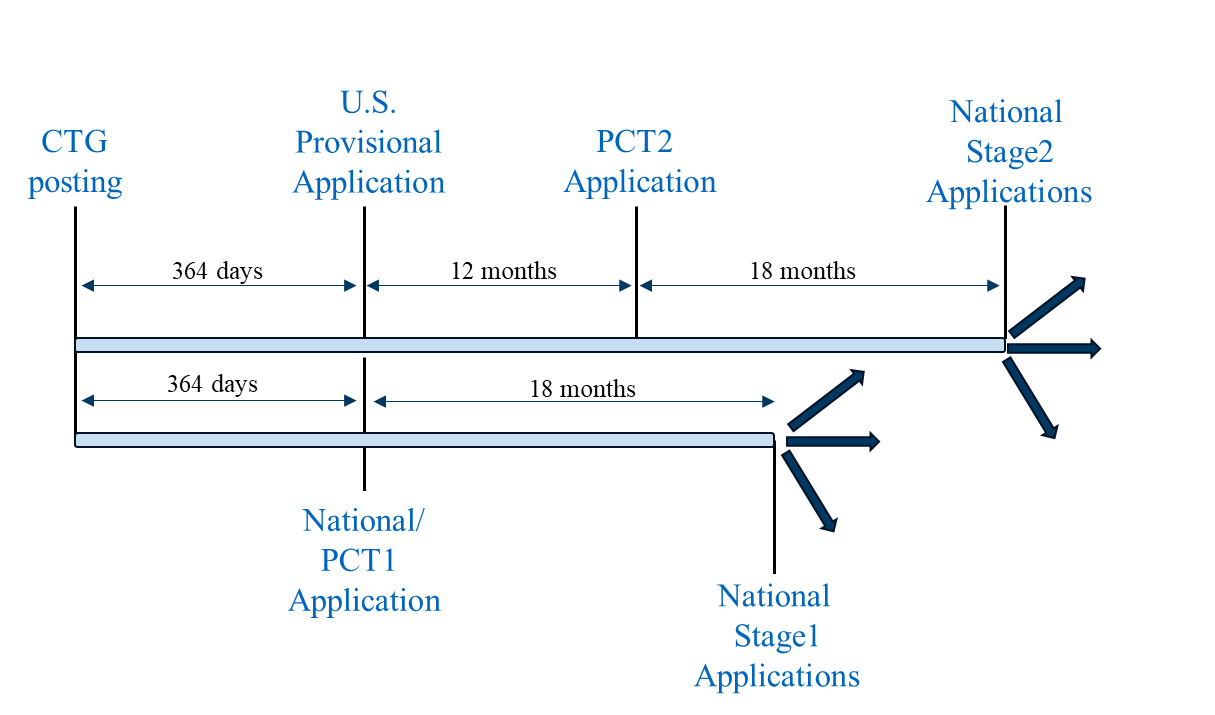
Scénario de classement 3
Une deuxième demande PCT (PCT2) revendiquant la priorité de la demande provisoire américaine peut alors être déposée à la date de la convention de 12 mois pour la demande provisoire américaine, qui comprend les résultats de l'essai clinique, s'ils sont disponibles. La demande PCT2 peut être nationalisée dans les juridictions où les exigences en matière de délai de grâce sont plus souples, comme indiqué ci-dessus pour le scénario 1.
Cette approche sur mesure maximise les avantages potentiels des délais de grâce, lorsqu'ils sont disponibles, tout en tenant compte de l'inclusion des données issues des essais cliniques (soit dans le PCT2, soit sous forme de données post-dépôt lors de la procédure nationale d'examen de la demande). Il en résulte une amélioration des chances d'obtenir la couverture de la méthode de traitement et une prolongation de la durée du brevet.
IV. Conclusion
La publication par le CTG d'un rapport d'étude de phase II ou III ou d'une divulgation publique connexe n'exclut pas nécessairement la brevetabilité des revendications relatives à un procédé de traitement dans une demande de brevet déposée ultérieurement sur la base du protocole d'essai clinique. Un nombre surprenant de juridictions accordent des délais de grâce pendant lesquels un rapport d'étude du CTG ou une divulgation connexe peuvent être retirés de l'état de la technique si une demande de brevet est déposée dans l'année suivant la divulgation. Cependant, le type de demande de brevet qui doit être déposé dans un délai d'un an varie selon les juridictions, ce qui conduit à des stratégies de dépôt complexes. Aux États-Unis, pour exclure les rapports d'étude du CTG en utilisant l'exception prévue à l'article 102(b)(1)(A), les praticiens doivent rédiger avec soin des déclarations conformes à la règle 130 qui établissent sans ambiguïté un lien entre le ou les inventeurs de la demande de brevet et le promoteur et le ou les chercheurs principaux de l'essai clinique. De même, les déclarations au titre de la règle 130 visant à exclure les divulgations liées aux essais cliniques en utilisant l'exception prévue à l'article 102(b)(1)(A) doivent expliquer sans ambiguïté toutes les différences entre le ou les auteurs de la divulgation et le ou les inventeurs de l'objet revendiqué.
[1] La plupart des juridictions hors des États-Unis n'autorisent pas les revendications relatives à la « méthode de traitement » en tant que telles, bien que ces revendications puissent être reformulées conformément aux pratiques locales, par exemple sous forme de revendications de type suisse et/ou de composés à usage limité pour les revendications de type d'utilisation.
[2] 42 U.S.C. § 282(j)(2)(A)
[3] 42 U.S.C. § 282(j)(2)(C)
[4] 42 U.S.C. § 282(j)(2)(D)
[5] Les modifications apportées à l'essai clinique, y compris les modifications du protocole, sont également publiées sur CTG selon le même mécanisme, de sorte que plusieurs dossiers d'étude sont généralement disponibles sur CTG pour un essai clinique donné.
[6] Voir, par exemple, Sanofi c. Glenmark Pharms, Inc., États-Unis, 204 F. Supp. 3d 665 (D. Del. 2016), confirmé sous le nom Sanofi c. Watson Lab’ys Inc., 875 F.3d 636 (Fed. Cir. 2017) (un brevet portant sur une méthode d'utilisation de la dronédarone dans le traitement de patients n'était pas prêt à être breveté avant la date critique et ne constituait donc pas un usage public) ; Dans l'affaire Omeprazole Patent Litigation, 536 F.3d 1361 (Fed. Cir. 2008) (un essai clinique de phase III ne constituait pas un usage public car l'invention n'avait pas été mise en pratique et n'était donc pas prête à être brevetée).
[7] IPR2023-01064, document 9 (P.T.A.B., 16 janvier 2024).
[8] Id., page 54.
[9] Id., page 55.
[10] Id., page 56.
[11] T 158/96, T 715/03, T 1859/08 et T 2506/12
[12] Dr Holger Tostmann, Bulletin d'information du Comité de pratique chimique de l'AIPLA, printemps 2024, volume 12, numéro I, p. 24.
[13] Au Canada, l'utilité est établie par démonstration ou « prédiction raisonnable » au moment où la demande est déposée.
