



Seeking Grace: Pursuing Method of Treatment Claims in View of Clinical Trial Related Disclosures

This article was originally published in the AIPLA Chemical Practice Chronicles Fall 2025 newsletter and is republished here with permission.
I. Background
Method of treatment patents based on Phase II and Phase III clinical trial protocols are routinely pursued to extend patent exclusivity and strategically build a patent portfolio for a drug asset. The claims of these “later-generation” method of treatment patents recite salient features of the Phase II or Phase III clinical trial protocol including patient populations, dosage amounts, dosing regimens, and efficacy outcome measurements. This is done for good reason, as the salient features of the study protocol often appear on the drug label, sometimes as part of explicit active steps.
For a new chemical entity (NCE), a later-generation method of treatment patent provides additional patent term, sometimes years beyond the patent term of the earlier-generation patents, i.e., foundational patents providing composition of matter and/or broad method of treatment exclusivity.
For repurposed drugs or novel dosing protocols, method of treatment patents based on Phase II or Phase III clinical trial study protocols may provide the only meaningful source of patent exclusivity, e.g., if the compound is known and the composition of matter patent has expired or is soon to expire.
Conventional wisdom dictates filing a patent application based on a Phase II or Phase III clinical trial protocol prior to a public disclosure of the study to avoid creation of prior art that can preclude patentability of the method of treatment claims, particularly outside the U.S.[1] Given the strategic importance of later-generation method of treatment patents, care should be taken to understand the timing of any public disclosures relating to the clinical trial and to plan patent application filings accordingly.
One such clinical trial related public disclosure is the posting of the innovator’s clinical trial protocol to ClinicalTrials.gov (“CTG”) as a so-called “study record.”[2] Posting of the study record is part of the U.S.’s well-intentioned effort to conduct human clinical trials with transparency to, inter alia, build public trust in clinical research and help patients find trials for which they might be eligible to participate. Innovators are to submit clinical trial study protocols to FDA no later than 21 days after the first patient is enrolled in the trial[3], and the study record will be posted not later than 30 days after submission[4][5]. Given these deadlines, it is typically not possible to predict the exact day a study record becomes public on CTG. Such an “un-docketable” deadline, unlike the firm date of an expected journal article publication or public symposium, has – perhaps unsurprisingly – led to a fact pattern where a study record is posted to CTG before a corresponding method of treatment patent application is filed.
II. Use of the § 102(b)(1)(A) exception
Clinical trials themselves are not considered a prior public use under 35 U.S.C. § 102(a)(1).[6] CTG study records and other clinical trial related disclosures, however, are considered printed publications and/or “otherwise available to the public” under §§ 102(a)(1) if made before the effective filing date of the patent application. Such clinical trial related disclosures can therefore be used to reject a later-filed patent application’s method of treatment claims as anticipated under 35 U.S.C. § 102(a)(1) and/or obvious under 35 U.S.C. § 103.
In the event a CTG study record or related disclosure becomes publicly available prior to filing a corresponding U.S. method of treatment patent application, the grace period exception under § 102(b)(1)(A) may be invoked to disqualify the disclosure under § 102(a)(1) – provided the disclosure is made one year or less before the effective filing date of the application. Procedurally, should an Examiner make a rejection under 35 U.S.C. §§ 102(a)(1) and/or 103 based on a CTG study record or related disclosure, Applicant can invoke the grace period by submitting a declaration under 37 CFR § 1.130 (“Rule 130”) establishing that the disclosure was made by the inventor or joint inventors and requesting removal of the CTG study record or related disclosure as prior art.
While submission of the Rule 130 declaration seems straightforward, a recent PTAB example, Murray & Poole Enterprises Ltd. v. Institut de Cardiologie de Montreal[7] is illustrative of potential traps. Institut de Cardiologie de Montreal (ICM) attempted to remove “Bouabdallaoui” as prior art by invoking the grace period exception under § 102(b)(1)(A). Bouabdallaoui was published within the one-year grace period and expanded on the results of the “COLCOT” CTG study record. Bouabdallaoui listed seven authors, the second of whom, Dr. Tardif, was the sole inventor on ICM’s patent. The board articulated that invocation of the grace period hinged on whether the declaration of Dr. Tardif provided sufficient information to conclude that Bouabdallaoui was not “by another.”[8] Dr. Tardif’s declaration explained the working relationship with only one co-author (Bouabdallaoui), as inter alia, acknowledgement of Bouabdallaoui’s assistance in conducting the clinical trial and to provide a first author publication. Dr. Tardif’s declaration also described the scope of the COLCOT multi-center clinical trial and ICM’s status as the sponsor, supported by agreements between some of the centers and the sponsor. The board pointed out that only a subset of center-sponsor agreements was provided and, among the agreements provided, none listed Dr. Tardif as the principal investigator.[9] Ultimately, in its decision of institution, the board found that Dr. Tardif’s declaration was insufficient to disqualify Bouabdallaoui as prior art.[10]
As Murray demonstrates, all differences between the authors of the clinical trial related disclosure and inventors on the patent application should be thoroughly explained. Declarations from any superfluous authors, i.e., non-inventors, disclaiming their contribution to the subject matter relied on in making the prior art rejection may help establish that the clinical trial related disclosure is not “by another.” CTG study records, unlike typical publications, list only the sponsor of a clinical trial, not specific individuals responsible for designing the study. Nonetheless, the inventor(s) can attest to the CTG study record as their own work in a declaration. Supporting documentation clearly tying the inventor(s) to the CTG study record, e.g., by naming the inventor(s) as the principal investigator(s), can also be provided.
III. International grace period provisions and filing strategies
In addition to the U.S., several foreign jurisdictions also provide a grace period. While not exhaustive, Table 1 summarizes grace period availability in commonly filed foreign jurisdictions, application filing timing, and whether proactive steps should be taken to make use of the grace period. Practitioners should work closely with local counsel to understand nuanced national requirements and timing to properly rely on each country’s available grace period.
Table 1
Country | Grace Period Available | Time to file application (from disclosure) | Type of first application that should be filed | Can the first application serve as priority document…† | Notes |
|---|---|---|---|---|---|
AU | Yes | 12 months | AU Standard Application or PCT | Yes (for PCT) | No proactive steps required. |
BR | Yes | 12 months | U.S. Provisional, BR, or PCT | Yes | No proactive steps required. |
CA | Yes | 12 months | CA or PCT | No | No proactive steps required. |
CN | No | — | — |
| CTG disclosure does not qualify for the grace period. |
EP | No | — | — |
| CTG disclosure does not qualify for the grace period. |
JP | Yes | 12 months | JP or PCT | No | Within 30 days of filing the JP national application or JP national phase entry, a certificate must be filed describing several features of the public disclosure. |
KR | Yes | 12 months | KR or PCT | Yes, but only if priority filing is (a) a PCT application designating only KR or (b) a direct KR application | Evidentiary documents must be submitted proving the applicant/inventor’s disclosure and showing (i) the date and type of the disclosure, (ii) the disclosing party, and (iii) the content of the disclosure. |
MX | Yes | 12 months | U.S. Provisional, MX, or PCT | Yes | The date of the public disclosure must be included on a form when filing the MX national application or MX national phase entry of the PCT. |
PH | Yes | 12 months | U.S. Provisional, PH, or PCT | Yes | No proactive steps required. |
SG | Yes | 12 months | PCT | No | No proactive steps required. |
TW | Yes | 12 months | TW | No | No proactive steps required. |
U.S. | Yes | 12 months | U.S. Provisional or PCT | Yes | No proactive steps required. |
† for a PCT or other application filed after the grace period that benefits from the grace period?
Like the U.S., Australia, Brazil, Canada, Japan, Mexico, the Philippines, South Korea, Singapore, and Taiwan all provide grace periods for CTG study records and related disclosures within 12 months of a first patent application’s filing date. They differ, however, with respect to whether the first patent application can serve as a priority document to a later-filed application, e.g., a PCT application, where the national stage application of the PCT application will also be eligible to benefit from the grace period. Certain jurisdictions, e.g., China and Europe, effectively do not have grace periods applicable to CTG study records or related disclosures.
In view of these distinctions, one can envisage complex potential filing strategies depending on international filing priorities.
Scenario 1 is a “typical” filing strategy where a U.S. provisional application is the first-filed application in the family. This filing strategy can be followed to pursue patent coverage in countries where a U.S. provisional application filed within 12 months of the CTG study record or related disclosure can serve as a priority document to a PCT application filed 12 months after the U.S. provisional application, and the national phase applications of the PCT application are eligible to benefit from the grace period based on the U.S. provisional application’s filing date. Representative countries in which this filing strategy can be utilized include the U.S., Brazil, Mexico, and the Philippines.
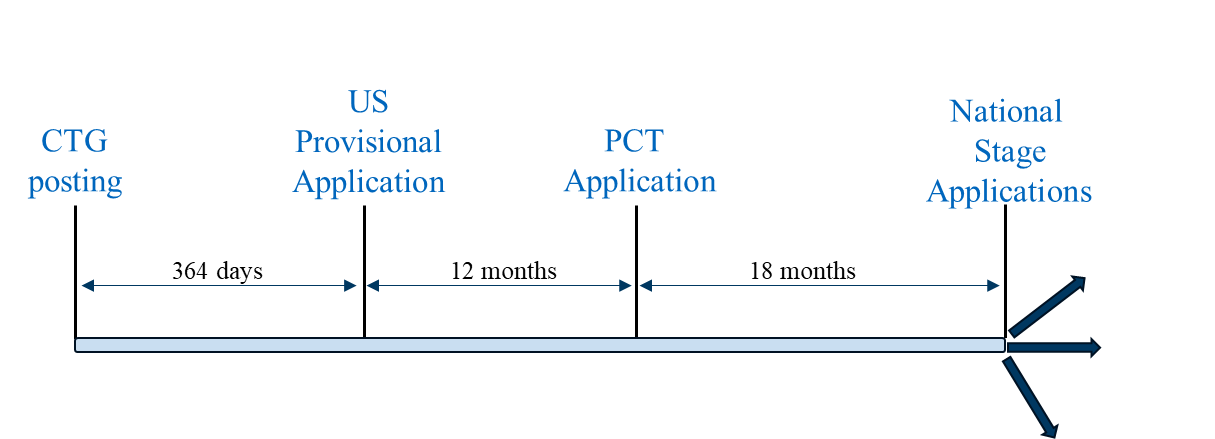
Filing Scenario 1
Scenario 1 can also be followed in countries that actually or effectively lack grace periods (e.g., China and Europe). To overcome the absolute novelty bar in China, possible aspects of the planned commercial methods that are not disclosed in the clinical trial related disclosure can be included in the U.S. provisional and/or PCT application. In Europe, CTG study records or announcements regarding ongoing trials not considered to be novelty destroying, although they can be used in an inventive step analysis.[11] For guidance on the implications of clinical trials as prior art in Europe, we direct the reader to the Spring 2024 AIPLA Chemical Practice Committee’s informative article on this very topic.[12]
Scenario 2 can be followed in countries where a PCT or non-PCT country national application must be filed within one year of the public disclosure for the national application to be eligible to benefit from a grace period. Representative countries in which this strategy can be utilized include Australia, Canada, Japan, Korea, Singapore, and Taiwan.
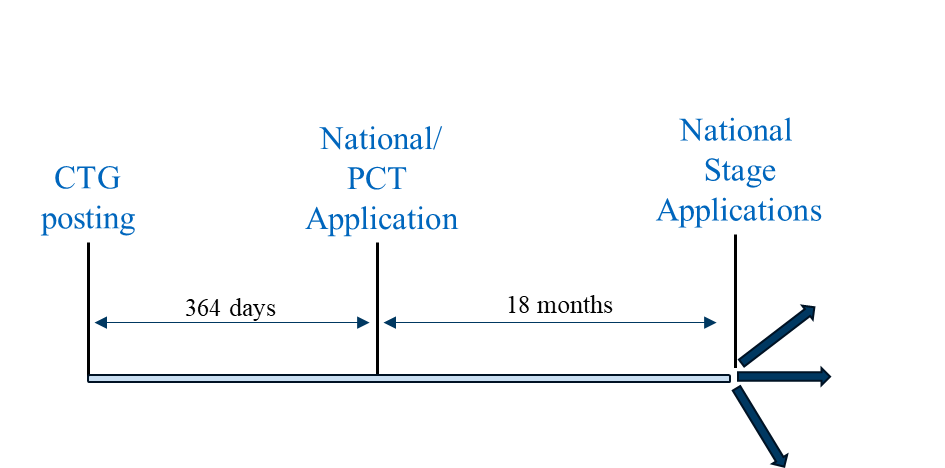
Filing Scenario 2
Scenario 1 provides two potential advantages compared to (a) a more conservative filing strategy where the U.S. provisional application is filed prior to the CTG study record posting and (b) Scenario 2: (1) maximum time for generating clinical trial results which, if available, can be included in the PCT application, and (2) the potential for maximum patent term.
Scenario 2 does not offer the benefit of an extended duration between CTG study record posting and national or PCT application filing. As such, results of the clinical trial are generally less likely to be available by the time a PCT application or non-PCT country national application is filed. However, many of the above-mentioned countries permit post-filing data during prosecution by which clinical trial results can be presented.[13]
Scenario 3 combines Scenario 1 and Scenario 2 into a unified filing strategy. First, a U.S. provisional application (following Scenario 1), a first PCT application (PCT1) (following Scenario 2), and non-PCT country national application(s) (following Scenario 2) are filed on the same day and within one year of the CTG study record posting. Prior to filing any applications outside the U.S., a foreign filing license should be considered and, if necessary, obtained. PCT1 can be nationalized in the jurisdictions with stricter grace period requirements outlined above for Scenario 2.
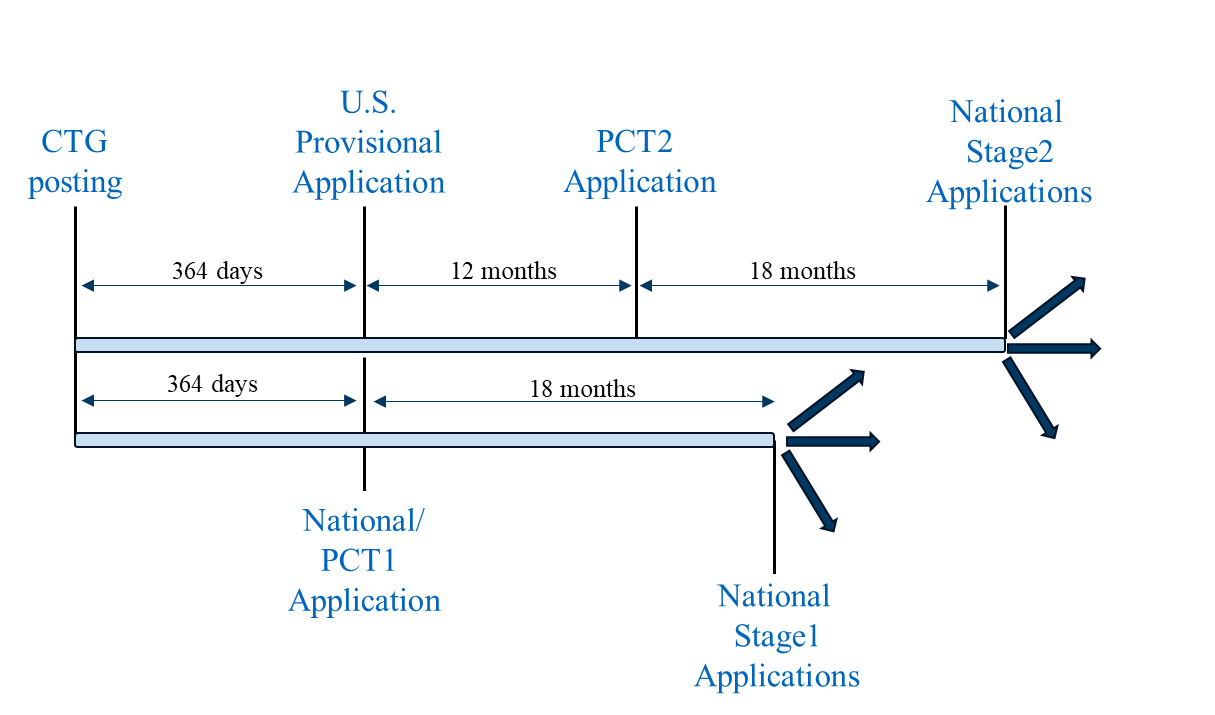
Filing Scenario 3
A second PCT application (PCT2) claiming priority to the U.S. provisional application can then be filed on the 12-month Convention date for the U.S. provisional application that includes the results of the clinical trial, if available. PCT2 can be nationalized in the jurisdictions with more relaxed grace period requirements as outlined above for Scenario 1.
This bespoke approach maximizes the potential benefits of grace periods, where available, while accounting for inclusion of clinical trial results data (either in PCT2 or as post-filing data during national stage application prosecution). The result is improved chances of obtaining method of treatment coverage and longer patent term.
IV. Conclusion
CTG posting of a Phase II or Phase III study record or a related public disclosure does not inevitably preclude patentability of method of treatment claims in a later-filed patent application based on the clinical trial protocol. A surprising number of jurisdictions provide grace periods by which a CTG study record or related disclosure can be removed as prior art if a patent application is filed within one year of the disclosure. However, the type of patent application that must be filed within one year varies by jurisdiction, thereby leading to complex filing strategies. In the U.S., to exclude CTG study records using the § 102(b)(1)(A) exception, practitioners should carefully draft Rule 130 declarations that unambiguously tie the inventor(s) of the patent application to the sponsor and principal investigator(s) of the clinical trial. Similarly, Rule 130 declarations to exclude clinical trial related disclosures using the § 102(b)(1)(A) exception should unambiguously explain any and all differences between the author(s) of the disclosure and the inventor(s) of the claimed subject matter.
[1] Most ex-US jurisdictions do not permit “method of treatment” claims per se, although such claims can be reformulated in accordance with local practice, e.g., as Swiss-type claims and/or purpose-limited compounds for use-type claims.
[2] 42 U.S.C. § 282(j)(2)(A)
[3] 42 U.S.C. § 282(j)(2)(C)
[4] 42 U.S.C. § 282(j)(2)(D)
[5] Changes to the clinical trial, including changes to the protocol, are also posted to CTG by the same mechanism such that multiple study records are typically available on CTG for a given clinical trial.
[6] See, e.g., Sanofi v. Glenmark Pharms, Inc., USA, 204 F. Supp. 3d 665 (D. Del. 2016), aff’d sub nom., Sanofi v. Watson Lab’ys Inc., 875 F.3d 636 (Fed. Cir. 2017) (A patent on a method of using dronedarone in treating patients was not ready for patenting before the critical date and thus not a public use); In Re Omeprazole Patent Litigation, 536 F.3d 1361 (Fed. Cir. 2008) (A Phase III clinical trial was not a public use because the invention had not been reduced to practice and therefore was not ready for patenting).
[7] IPR2023-01064, Paper 9 (P.T.A.B., Jan. 16, 2024).
[8] Id., page 54.
[9] Id., page 55.
[10] Id., page 56.
[11] T 158/96, T 715/03, T 1859/08 and T 2506/12
[12] Dr. Holger Tostmann, Newsletter of the AIPLA Chemical Practice Committee, Spring 2024, Volume 12, Issue I, p. 24.
[13] In Canada, utility is established by demonstration or “sound prediction” at the time the application is filed.
