



En busca de la gracia: reclamación de métodos de tratamiento a la luz de las revelaciones relacionadas con ensayos clínicos
Este artículo se publicó originalmente en Boletín de Práctica Química de la AIPLA, otoño de 2025 y se reproduce aquí con permiso.
I. Antecedentes
Las patentes de métodos de tratamiento basadas en protocolos de ensayos clínicos de fase II y fase III se solicitan de forma rutinaria para ampliar la exclusividad de la patente y crear estratégicamente una cartera de patentes para un activo farmacéutico. Las reivindicaciones de estas patentes de métodos de tratamiento de «última generación» recogen las características más destacadas del protocolo de ensayos clínicos de fase II o fase III, incluyendo las poblaciones de pacientes, las dosis, los regímenes de dosificación y las mediciones de los resultados de eficacia. Esto se hace por una buena razón, ya que las características más destacadas del protocolo del estudio suelen aparecer en la etiqueta del medicamento, a veces como parte de pasos activos explícitos.
En el caso de una nueva entidad química (NCE), una patente de método de tratamiento de última generación proporciona un plazo de patente adicional, a veces años más allá del plazo de las patentes de generación anterior, es decir, las patentes fundamentales que proporcionan la composición de la materia y/o la exclusividad del método de tratamiento en general.
En el caso de los medicamentos reutilizados o los nuevos protocolos de dosificación, las patentes de métodos de tratamiento basadas en protocolos de ensayos clínicos de fase II o fase III pueden proporcionar la única fuente significativa de exclusividad de la patente, por ejemplo, si el compuesto es conocido y la patente de composición de la materia ha expirado o está a punto de hacerlo.
La sabiduría convencional dicta presentar una solicitud de patente basada en un protocolo de ensayo clínico de fase II o fase III antes de la divulgación pública del estudio, con el fin de evitar la creación de antecedentes que puedan impedir la patentabilidad de las reivindicaciones del método de tratamiento, especialmente fuera de los Estados Unidos.[1] Dada la importancia estratégica de las patentes de métodos de tratamiento de última generación, se debe prestar especial atención a comprender el momento adecuado para cualquier divulgación pública relacionada con el ensayo clínico y planificar la presentación de solicitudes de patente en consecuencia.
Una de estas divulgaciones públicas relacionadas con ensayos clínicos es la publicación del protocolo del ensayo clínico del innovador en ClinicalTrials.gov («CTG») como un «registro de estudio».[2] La publicación del registro del estudio forma parte del esfuerzo bienintencionado de los Estados Unidos por llevar a cabo ensayos clínicos en seres humanos con transparencia, entre otras cosas, generar confianza pública en la investigación clínica y ayudar a los pacientes a encontrar ensayos en los que puedan participar. Los innovadores deben presentar los protocolos de los ensayos clínicos a la FDA a más tardar 21 días después de que el primer paciente se inscriba en el ensayo[3], y el registro del estudio se publicará a más tardar 30 días después de la presentación[4][5]. Dados estos plazos, normalmente no es posible predecir el día exacto en que un registro de estudio se hará público en el CTG. Este plazo «no registrable», a diferencia de la fecha fija de publicación de un artículo en una revista o de un simposio público, ha dado lugar, como era de esperar, a una situación en la que el registro de un estudio se publica en el CTG antes de que se presente la correspondiente solicitud de patente del método de tratamiento.
II. Uso de la excepción del § 102(b)(1)(A)
Los ensayos clínicos en sí mismos no se consideran un uso público previo según lo establecido en el artículo 35 U.S.C. § 102(a)(1).[6] Sin embargo, los registros de estudios de CTG y otras divulgaciones relacionadas con ensayos clínicos se consideran publicaciones impresas y/o «disponibles de otro modo para el público» en virtud de los §§ 102(a)(1) si se realizan antes de la fecha efectiva de presentación de la solicitud de patente. Por lo tanto, dichas divulgaciones relacionadas con ensayos clínicos pueden utilizarse para rechazar las reivindicaciones de métodos de tratamiento de una solicitud de patente presentada posteriormente como anticipadas en virtud del artículo 35 U.S.C. § 102(a)(1) y/o obvias en virtud del artículo 35 U.S.C. § 103.
En caso de que un registro de estudio CTG o una divulgación relacionada se haga público antes de presentar la correspondiente solicitud de patente de método de tratamiento en EE. UU., se podrá invocar la excepción del período de gracia prevista en el artículo 102(b)(1)(A) para descalificar la divulgación en virtud del artículo 102(a)(1), siempre que la divulgación se haya realizado un año o menos antes de la fecha efectiva de presentación de la solicitud. Desde el punto de vista procedimental, si un examinador rechaza una solicitud en virtud de los artículos 35 U.S.C. §§ 102(a)(1) y/o 103 basándose en un registro de estudio de CTG o una divulgación relacionada, el solicitante puede invocar el período de gracia presentando una declaración en virtud del artículo 37 CFR § 1.130 («Regla 130») en la que se establezca que la divulgación fue realizada por el inventor o los coinventores y se solicite la eliminación del registro del estudio del CTG o la divulgación relacionada como estado de la técnica.
Aunque la presentación de la declaración de la Regla 130 parece sencilla, un ejemplo reciente de la PTAB, Murray & Poole Enterprises Ltd. contra Institut de Cardiologie de Montreal[7] ilustra las posibles trampas. El Institut de Cardiologie de Montreal (ICM) intentó eliminar «Bouabdallaoui» como estado de la técnica invocando la excepción del período de gracia en virtud del artículo 102(b)(1)(A). Bouabdallaoui se publicó dentro del período de gracia de un año y amplió los resultados del estudio CTG «COLCOT». Bouabdallaoui enumeraba siete autores, el segundo de los cuales, el Dr. Tardif, era el único inventor de la patente del ICM. La junta articuló que la invocación del período de gracia dependía de si la declaración del Dr. Tardif proporcionaba información suficiente para concluir que Bouabdallaoui no era «de otro».[8] La declaración del Dr. Tardif explicaba la relación laboral únicamente con un coautor (Bouabdallaoui), como inter aliael reconocimiento de la ayuda de Bouabdallaoui en la realización del ensayo clínico y para proporcionar una publicación como primer autor. La declaración del Dr. Tardif también describía el alcance del ensayo clínico multicéntrico COLCOT y la condición de ICM como patrocinador, respaldada por acuerdos entre algunos de los centros y el patrocinador. La junta señaló que solo se proporcionó un subconjunto de acuerdos entre los centros y el patrocinador y que, entre los acuerdos proporcionados, ninguno incluía al Dr. Tardif como investigador principal.[9] En última instancia, en su decisión de institución, la junta consideró que la declaración del Dr. Tardif era insuficiente para descalificar a Bouabdallaoui como estado de la técnica.[10]
Como demuestra Murray , todas las diferencias entre los autores de la divulgación relacionada con el ensayo clínico y los inventores de la solicitud de patente deben explicarse detalladamente. Las declaraciones de cualquier autor superfluo, es decir, que no sea inventor, en las que renuncie a su contribución al tema en el que se basa el rechazo por estado de la técnica, pueden ayudar a establecer que la divulgación relacionada con el ensayo clínico no es «de otro». Los registros de estudios del CTG, a diferencia de las publicaciones típicas, solo incluyen al patrocinador de un ensayo clínico, y no a las personas específicas responsables del diseño del estudio. No obstante, los inventores pueden dar fe del registro del estudio del CTG como su propio trabajo en una declaración. También se puede proporcionar documentación de apoyo que vincule claramente a los inventores con el registro del estudio del CTG, por ejemplo, nombrando a los inventores como investigadores principales.
III. Disposiciones internacionales sobre el período de gracia y estrategias de presentación
Además de los Estados Unidos, varias jurisdicciones extranjeras también ofrecen un período de gracia. Aunque no es exhaustiva, la Tabla 1 resume la disponibilidad del período de gracia en las jurisdicciones extranjeras más comunes, los plazos para la presentación de solicitudes y si se deben tomar medidas proactivas para aprovechar el período de gracia. Los profesionales deben trabajar en estrecha colaboración con los abogados locales para comprender los requisitos nacionales y los plazos con el fin de aprovechar adecuadamente el período de gracia disponible en cada país.
Tabla 1
País | Período de gracia disponible | Plazo para presentar la solicitud (desde la divulgación) | Tipo de primera solicitud que debe presentarse | ¿Puede la primera solicitud servir comodocumento prioritario…? | Notas |
|---|---|---|---|---|---|
UA | Sí | 12 meses | Solicitud estándar de la UA o PCT | Sí (para PCT) | No se requieren medidas proactivas. |
BR | Sí | 12 meses | Provisional de EE. UU., BR o PCT | Sí | No se requieren medidas proactivas. |
CA | Sí | 12 meses | CA o PCT | No | No se requieren medidas proactivas. |
CN | No | — | — |
| La divulgación de CTG no cumple los requisitos para el período de gracia. |
EP | No | — | — |
| La divulgación de CTG no cumple los requisitos para el período de gracia. |
JP | Sí | 12 meses | JP o PCT | No | En un plazo de 30 días a partir de la presentación de la solicitud nacional JP o de la entrada en la fase nacional JP, se debe presentar un certificado en el que se describan varias características de la divulgación pública. |
KR | Sí | 12 meses | KR o PCT | Sí, pero solo si la presentación prioritaria es (a) una solicitud PCT que designa únicamente a Corea o (b) una solicitud directa a Corea. | Se deben presentar documentos probatorios que demuestren la divulgación del solicitante/inventor y que indiquen (i) la fecha y el tipo de divulgación, (ii) la parte divulgadora y (iii) el contenido de la divulgación. |
MX | Sí | 12 meses | Provisional de EE. UU., MX o PCT | Sí | La fecha de la divulgación pública debe incluirse en un formulario al presentar la solicitud nacional MX o la entrada en la fase nacional MX del PCT. |
PH | Sí | 12 meses | Provisional de EE. UU., PH o PCT | Sí | No se requieren medidas proactivas. |
SG | Sí | 12 meses | PCT | No | No se requieren medidas proactivas. |
TW | Sí | 12 meses | TW | No | No se requieren medidas proactivas. |
EE. UU. | Sí | 12 meses | Provisional de EE. UU. o PCT | Sí | No se requieren medidas proactivas. |
† ¿Para una solicitud PCT u otra solicitud presentada después del período de gracia que se beneficia del período de gracia?
Al igual que Estados Unidos, Australia, Brasil, Canadá, Japón, México, Filipinas, Corea del Sur, Singapur y Taiwán ofrecen períodos de gracia para los registros de estudios de CTG y las divulgaciones relacionadas dentro de los 12 meses siguientes a la fecha de presentación de la primera solicitud de patente. Sin embargo, difieren en cuanto a si la primera solicitud de patente puede servir como documento de prioridad para una solicitud presentada posteriormente, por ejemplo, una solicitud PCT, en la que la solicitud en la fase nacional de la solicitud PCT también podrá beneficiarse del período de gracia. Algunas jurisdicciones, como China y Europa, no tienen períodos de gracia aplicables a los registros de estudios de CTG o divulgaciones relacionadas.
En vista de estas distinciones, se pueden prever complejas estrategias de presentación de solicitudes en función de las prioridades internacionales en materia de presentación.
El escenario 1 es una estrategia de presentación «típica» en la que una solicitud provisional estadounidense es la primera solicitud presentada en la familia. Esta estrategia de presentación puede seguirse para obtener la cobertura de la patente en países en los que una solicitud provisional estadounidense presentada en los 12 meses siguientes al registro del estudio del CTG o a la divulgación relacionada puede servir como documento de prioridad para una solicitud PCT presentada 12 meses después de la solicitud provisional estadounidense, y las solicitudes de la fase nacional de la solicitud PCT pueden beneficiarse del período de gracia basado en la fecha de presentación de la solicitud provisional estadounidense. Entre los países representativos en los que se puede utilizar esta estrategia de presentación se incluyen Estados Unidos, Brasil, México y Filipinas.
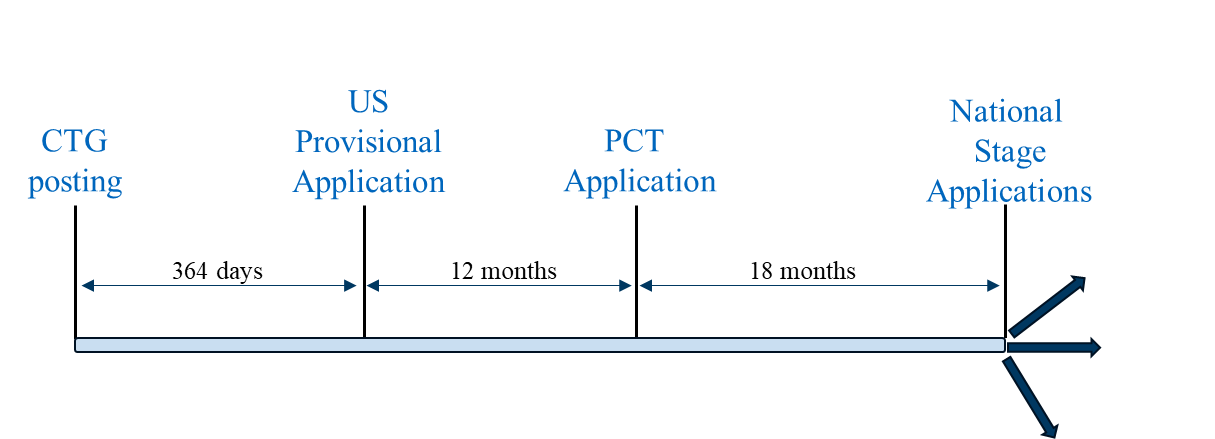
Escenario de presentación 1
El escenario 1 también puede seguirse en países que carecen real o efectivamente de períodos de gracia (por ejemplo, China y Europa). Para superar la barrera de la novedad absoluta en China, los posibles aspectos de los métodos comerciales previstos que no se divulgan en la divulgación relacionada con los ensayos clínicos pueden incluirse en la solicitud provisional y/o PCT de los Estados Unidos. En Europa, los registros de estudios del CTG o los anuncios relativos a ensayos en curso no se consideran destructores de la novedad, aunque pueden utilizarse en un análisis de la actividad inventiva.[11] Para obtener orientación sobre las implicaciones de los ensayos clínicos como estado de la técnica en Europa, remitimos al lector al artículo informativo del Comité de Práctica Química de la AIPLA de la primavera de 2024 sobre este mismo tema.[12]
El escenario 2 puede seguirse en países en los que debe presentarse una solicitud nacional PCT o no PCT en el plazo de un año desde la divulgación pública para que la solicitud nacional pueda beneficiarse de un período de gracia. Entre los países representativos en los que puede utilizarse esta estrategia se incluyen Australia, Canadá, Japón, Corea, Singapur y Taiwán.
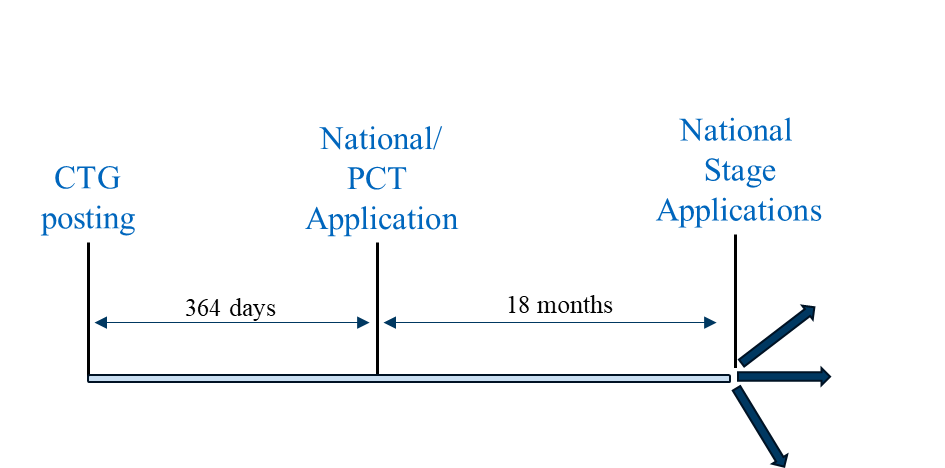
Escenario de presentación 2
El escenario 1 ofrece dos ventajas potenciales en comparación con (a) una estrategia de presentación más conservadora, en la que la solicitud provisional estadounidense se presenta antes de la publicación del registro del estudio CTG, y (b) el escenario 2: (1) el tiempo máximo para generar los resultados del ensayo clínico que, si están disponibles, pueden incluirse en la solicitud PCT, y (2) la posibilidad de obtener la duración máxima de la patente.
El escenario 2 no ofrece la ventaja de un plazo más amplio entre la publicación del registro del estudio CTG y la presentación de la solicitud nacional o PCT. Por lo tanto, es menos probable que los resultados del ensayo clínico estén disponibles en el momento de presentar una solicitud PCT o una solicitud nacional en un país no PCT. Sin embargo, muchos de los países mencionados anteriormente permiten presentar datos posteriores a la presentación durante el proceso de tramitación, lo que permite presentar los resultados de los ensayos clínicos.[13]
El escenario 3 combina el escenario 1 y el escenario 2 en una estrategia de presentación unificada. En primer lugar, se presenta una solicitud provisional en los Estados Unidos (siguiendo el escenario 1), una primera solicitud PCT (PCT1) (siguiendo el escenario 2) y una o varias solicitudes nacionales no PCT (siguiendo el escenario 2) el mismo día y en el plazo de un año desde la publicación del registro del estudio del CTG. Antes de presentar cualquier solicitud fuera de los Estados Unidos, se debe considerar la posibilidad de obtener una licencia de presentación en el extranjero y, si es necesario, obtenerla. La PCT1 puede nacionalizarse en las jurisdicciones con requisitos de plazo de gracia más estrictos, tal y como se describe anteriormente para el escenario 2.
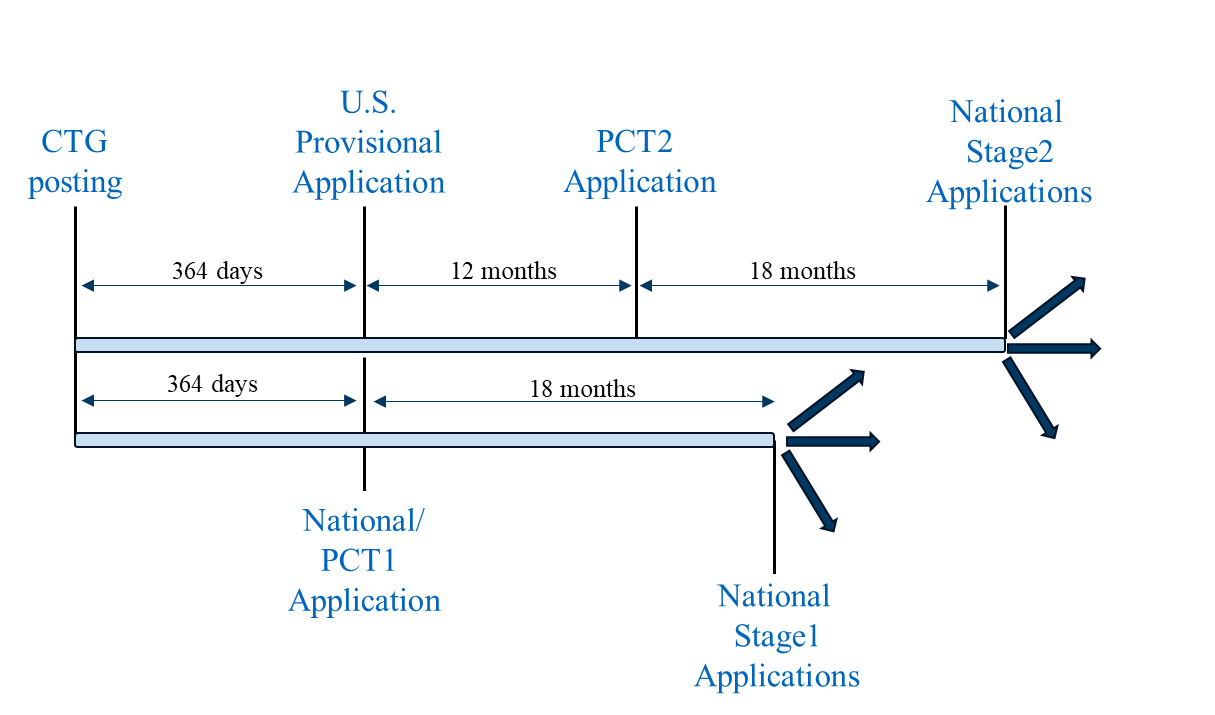
Escenario de presentación 3
A continuación, se puede presentar una segunda solicitud PCT (PCT2) que reivindique la prioridad de la solicitud provisional estadounidense en la fecha de la Convención de 12 meses para la solicitud provisional estadounidense, que incluya los resultados del ensayo clínico, si están disponibles. La PCT2 se puede nacionalizar en las jurisdicciones con requisitos de período de gracia más flexibles, tal y como se ha descrito anteriormente para el escenario 1.
Este enfoque personalizado maximiza los posibles beneficios de los períodos de gracia, cuando están disponibles, al tiempo que tiene en cuenta la inclusión de los datos de los resultados de los ensayos clínicos (ya sea en PCT2 o como datos posteriores a la presentación durante la tramitación de la solicitud en la fase nacional). El resultado es una mayor probabilidad de obtener la cobertura del método de tratamiento y una mayor duración de la patente.
IV. Conclusión
La publicación por parte del CTG de un registro de un estudio de fase II o fase III o una divulgación pública relacionada no excluye inevitablemente la patentabilidad de las reivindicaciones de métodos de tratamiento en una solicitud de patente presentada posteriormente basada en el protocolo del ensayo clínico. Un número sorprendente de jurisdicciones ofrecen períodos de gracia durante los cuales se puede eliminar como estado de la técnica un registro de estudio del CTG o una divulgación relacionada si la solicitud de patente se presenta en el plazo de un año desde la divulgación. Sin embargo, el tipo de solicitud de patente que debe presentarse en el plazo de un año varía según la jurisdicción, lo que da lugar a estrategias de presentación complejas. En los Estados Unidos, para excluir los registros de estudios del CTG utilizando la excepción del artículo 102(b)(1)(A), los profesionales deben redactar cuidadosamente las declaraciones de la Regla 130 que vinculen de forma inequívoca al inventor o inventores de la solicitud de patente con el patrocinador y los investigadores principales del ensayo clínico. Del mismo modo, las declaraciones de la Regla 130 para excluir las divulgaciones relacionadas con ensayos clínicos utilizando la excepción § 102(b)(1)(A) deben explicar sin ambigüedades todas y cada una de las diferencias entre el autor o autores de la divulgación y el inventor o inventores del objeto reivindicado.
[1] La mayoría de las jurisdicciones fuera de los Estados Unidos no permiten las reclamaciones por «método de tratamiento» per se, aunque dichas reivindicaciones pueden reformularse de acuerdo con la práctica local, por ejemplo, como reivindicaciones de tipo suizo y/o compuestos con fines limitados para reivindicaciones de tipo de uso.
[2] 42 U.S.C. § 282(j)(2)(A)
[3] 42 U.S.C. § 282(j)(2)(C)
[4] 42 U.S.C. § 282(j)(2)(D)
[5] Los cambios en el ensayo clínico, incluidos los cambios en el protocolo, también se publican en CTG mediante el mismo mecanismo, de modo que normalmente hay varios registros de estudio disponibles en CTG para un ensayo clínico determinado.
[6] Véase, por ejemplo, Sanofi contra Glenmark Pharms, Inc., EE. UU., 204 F. Supp. 3d 665 (D. Del. 2016), confirmado sub nom., Sanofi contra Watson Lab’ys Inc., 875 F.3d 636 (Fed. Cir. 2017) (una patente sobre un método de uso de dronedarona en el tratamiento de pacientes no estaba lista para ser patentada antes de la fecha crítica y, por lo tanto, no era de uso público); En el litigio sobre la patente del omeprazol, 536 F.3d 1361 (Fed. Cir. 2008) (un ensayo clínico de fase III no era un uso público porque la invención no se había puesto en práctica y, por lo tanto, no estaba lista para ser patentada).
[7] IPR2023-01064, Documento 9 (P.T.A.B., 16 de enero de 2024).
[8] Id., página 54.
[9] Id., página 55.
[10] Id., página 56.
[11] T 158/96, T 715/03, T 1859/08 y T 2506/12
[12] Dr. Holger Tostmann, Boletín informativo del Comité de Práctica Química de la AIPLA, primavera de 2024, volumen 12, número I, p. 24.
[13] En Canadá, la utilidad se establece mediante demostración o «predicción sólida» en el momento en que se presenta la solicitud.
